Reflection on my time working on Direct Atomics (deep tech startup) with Curtis (May-Sep 2024).
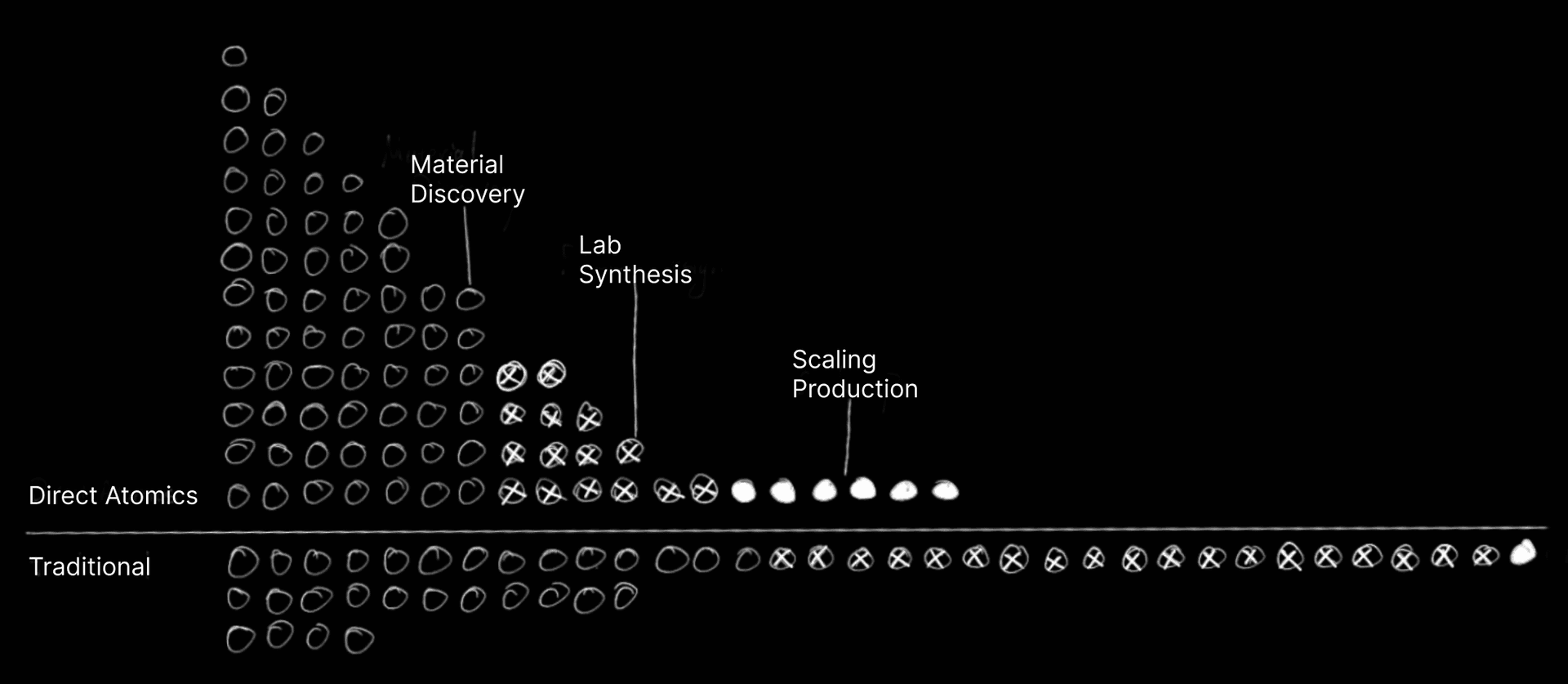
The Slow Science Problem
Imagine spending billions of dollars and waiting years to launch a new product-only to watch it flop. This isn't a worst-case scenario. It's business as usual for companies developing new materials.
Take the phone in your pocket. Its chips come from TSMC, where scientists spend years testing different atomic recipes. Or consider how BASF develops cleaner fuel catalysts-each one demanding countless experiments. Even Merck's life-saving drugs follow this painfully slow process. Why so slow? Because atoms are social. To simulate just 1,000 of them, a computer needs to track a million relationships. Add more atoms, and the problem explodes-like trying to map every conversation at an ever-growing party.
These atomic simulations drive modern innovation:
- NASA uses them to turn spacecraft heat into electricity
- Drug companies use them to design disease-fighting molecules
- Chip makers use them to build faster, more efficient processors
Cheating Atomic Math
Predicting how electrons move around atoms is one of physics' hardest problems. Each electron affects every other electron, leading to impossible calculations. Scientists solved this with a shortcut called Density Functional Theory (DFT). Instead of tracking individual electrons, DFT tracks where electrons are likely to be found. This simpler approach tells us enough about a material while making calculations possible. But it's still too slow for large problems with thousands of atoms. That's where neural networks come in. By learning from DFT calculations, these networks can predict how new atomic arrangements will behave almost as accurately but much faster.

Here's what that means: At 3,000 atoms, a neural network on a single GPU outperforms a 36-core supercomputer by 17.5 million times. What once took years now takes weeks. Million-dollar experiments become thousand-dollar ones. The difference isn't just speed-it's possibility. We can now simulate entire proteins, design materials atom by atom, and tackle problems that were previously unthinkable.
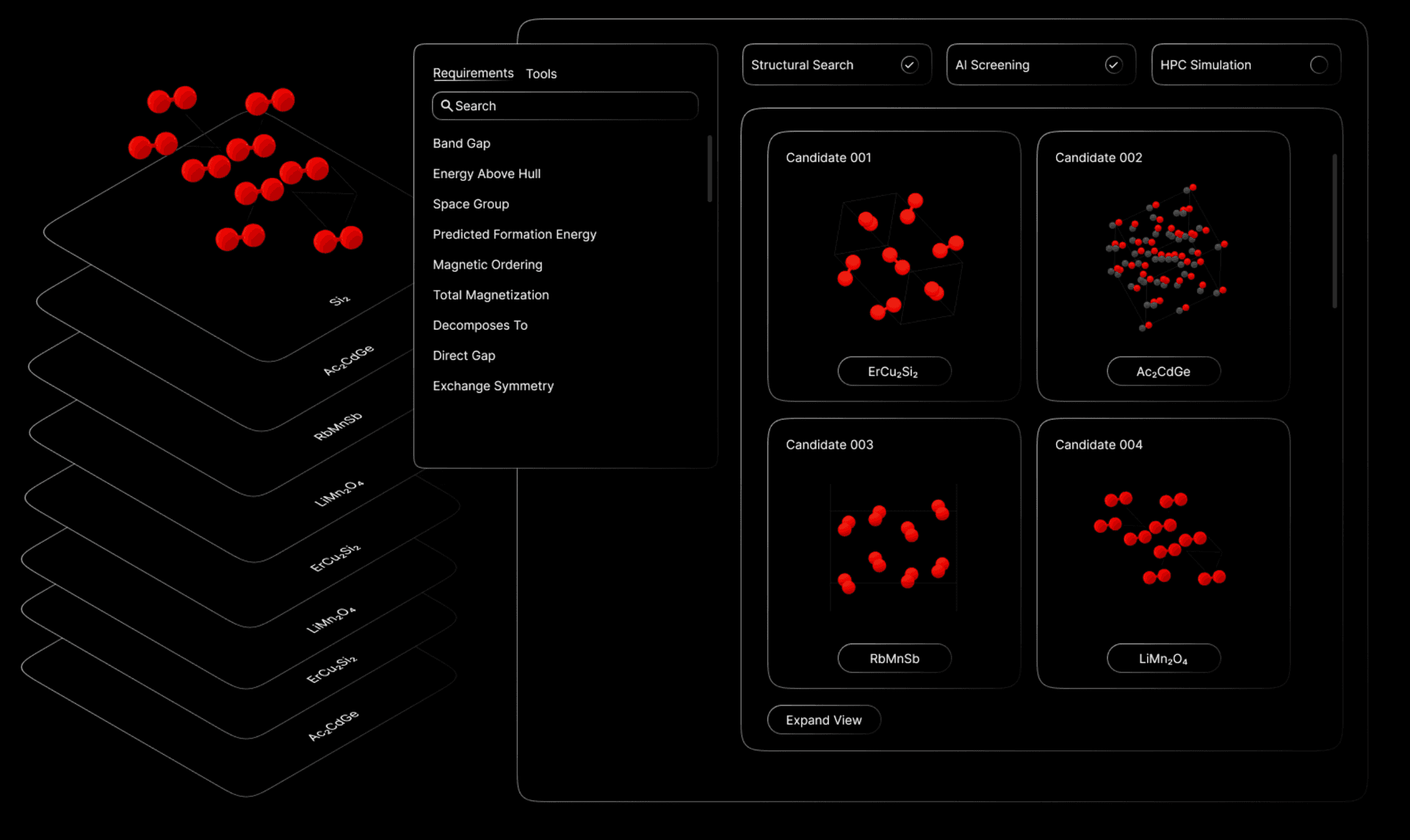
Click, Drop, Compute
The core technology is an Equivariant Graph Neural Network. The input is the atomic system (types, postitions of atoms, lattice sturcture), and the output is the system property like total energy or forces per atom.
In June, we trained a small model with 13.8 million parameters (332 MB) using the eSCN architecture on data from the top 20 most common elements from the Alexandria dataset. Our goal was to validate the training process on a manageable scale before moving to the full datasets (≈20 million), running it on a single GPU for 8 hours. This exercise revealed issues such as duplicate entries in the data and unexpectedly high force values. We resolved these by cleaning the dataset and implementing tests to verify the model’s accuracy, including checks for consistent outputs under specific transformations.

Quite a bit of effort also went into reimagined the user experience. Below is a UX comparsion between trandtional methods and Direct Atomics.
Traditional VASP Workflow (Months)
- Apply for a license (~$5.6k/seat)
- Install on a supercomputer
- Filter structures manually
- Prepare input files
- Apply for supercomputer time
- Upload files via CLI and wait in a queue
- Run the computation
- Interpret the results
Direct Atomics (Minutes)
- Sign up
- Upload structure files (drag and drop or CLI)
- Computation
- Interpret results
What Went Wrong?
A lot of things. In short, we made something that people don’t want.
What the heck, there are literal playbooks not to fumble like we did- Unusual VC’s Discovery’s Playbook, The Mom Test. Now as with anything, it's nuanced. But in a few sentences, here is the problem:
We started with the idea that computational tools could help industries screen materials faster and better. We made this decision based on what we saw in academia and national lab research.
In industries like semiconductors, material screening isn’t the bottleneck. There are billions of dollars invested in optimizing materials and processes to achieve precise, physical outcomes. If a computationalist finds a material that’s 20% better for a use case, nobody else in the company cares. It’s not just about finding a better material-it’s about everything else already in place.
 Oops.
Oops.Here are a few of the other influencing reasons we decided to pivot:
- Market size is too small: Only about 100 multi-million dollar contract customers = Long negotiated deals.
- Customers won’t use SOTA models: They rely on cheaper heuristics (element types, in-house models) to narrow search spaces, limiting revenue.
- Schrödinger's scaling problem: 30 years in, they generate $100M ARR, mostly from on-prem installs, with scaling hampered by heavy customization needs.
- Workflow pride: Computationalists see drag-and-drop interfaces as insulting; they prefer running specific simulations to understand material chemistry.
- In-house capabilities: Companies want to develop their models to avoid third-party reliance.
- Wealthy companies: Private companies that can pay are rich enough to run expensive physics calculations themselves.
- Open-source decentralization: Many functionalities are already decentralized due to open-source models.
You wouldn’t know these challenges unless have tried selling to industry customers.
Mistakes We Made
One of our best decisions was taking a day for a pre-mortem-writing down every critical assumption that had to hold true for our startup to succeed. No founder wants to do this; it’s like a parent listing every flaw of their child. But facing those uncomfortable truths helped us gather the learnings below and make the pivot we needed.
There is The Pocket Guide of Essential YC Advice which we initially thought didn’t apply to us because we were doing Deep Tech- spoiler alert it did:
-
We spent a month trying to train a diffusion model to generate new materials-turns out, we didn’t have enough data. Then, we tried training the model again-didn’t have the money. Another month down the drain. Don’t build stuff that would be useful someday -build for now.
-
If you’re proud of the first product you launched, you probably waited too long to launch. There’s always a 90/10 solution. In our case, the janky, fragile, non-scalable version was the one that worked. From an engineering standpoint-holy shit-it was horrible. But it got us traction.
-
Don’t scale your team until you’ve built something people want. We reached out to 200 ML and infrastructure engineers-algorithm-heavy talent from quant firms and academia-and convinced eight to join once we secured funding. We did this to impress investors and outpace the competition. In hindsight, private industry didn’t want the product. Scaling before knowing that was a waste of time.
-
Ignore your competitors-you’ll die of suicide, not murder. But we couldn’t help ourselves. Stalking them filled some domain expertise gaps and prevented us from getting grilled by VCs, but it also caused unnecessary stress. How were we supposed to outcompete Microsoft Quantum with an army of MIT PhDs? Or Matlantis with most of Asia’s Kaggle grandmasters? Or CUSP with Geoffrey Hinton? In reality, the problem space was already kicking our collective asses, Nobel Prize winner or not. Worrying about things we couldn’t control was a waste of time.
-
Talk to users first, then write code. You can’t learn or build everything. Reading papers and writing code is not a substitute for real user conversations.You never want to be in the potision of a technology searching for a problem. Assume you know nothing unless you’re the expert. Code should test specific, predefined hypotheses. We failed because of business issues, not technical ones, so focus on solving real-world problems by listening first.
Days to Pivot
Days before our second YC interview and investor calls, we realized Direct Atomics wasn’t venture-scale, though some investors remained interested. We needed a new idea. Here’s what we tried:
- Scoured Our Experiences: Looked for gaps in our work and personal lives-but only came up with ordinary ideas.
- Considered Recent World Changes: This led us to solutions searching for problems.
- Targeted Low NPS Giants: Identified big companies with poor customer satisfaction to improve their user experience.
- Talked to Uncommon Voices: Spoke with professionals outside tech-doctors, nurses-to gain fresh perspectives.
- Explored Online Epiphanies: Read about life-changing experiences on forums.
We rated each idea from 1 to 10 on:
- Opportunity Size
- Founder-Market Fit
- Ease of Starting
- Early Market Feedback
No new insights came from this, however. Ideas best happen organically.
The Technical
Density Functional Theory
-
The Many-Body Schrödinger Equation
The equation that describes N interacting electrons:
This equation governs the behavior of all N electrons in a system. It describes how their positions and energy are related, but it's impossible to solve as the number of electrons grows because we have to track too many interactions.
-
Hamiltonian Operator (Ĥ)
The Hamiltonian, which represents the total energy:
The Hamiltonian tells us the total energy of the system: the first term is the kinetic energy of the electrons, the second term is the potential energy due to external fields, and the third term is the electron-electron repulsion. Solving this for N electrons is complex because every electron interacts with every other one.
-
Why It Grows on 3N Coordinates?
The wavefunction depends on the positions of all electrons:
As the number of electrons (N) increases, the complexity explodes because the wavefunction depends on 3 coordinates (x, y, z) for every electron. That’s 3N dimensions to deal with, making it impossible to solve directly as N grows.
-
Density Functional Theory (DFT) Simplification
DFT replaces the wavefunction with the electron density :
DFT cuts down the complexity by using the electron density instead of the full wavefunction. This is like summarizing the entire system by saying, “Here’s how many electrons are at each point,” instead of tracking them all individually.
-
Kohn-Sham Equations
The Kohn-Sham equations describe how to compute the electron density:
This equation breaks the big problem into smaller pieces. Instead of solving one giant equation for all electrons, it treats each electron individually but with an effective potential that takes all interactions into account. It's like turning a huge puzzle into smaller, manageable chunks.
-
Iterative Process
The process DFT uses to find the correct electron density:
- Guess initial .
- Calculate the effective potential .
- Solve the Kohn-Sham equations for .
- Update the electron density .
- Repeat until it converges.
The TLDR is DFT works by guessing a solution, refining it, and repeating this process until everything fits together. It’s like iterating over guesses for the answer until it stops changing.
Equivariant GNNS
Equivariance ensures that the same compound, even when rotated differently, is seen as the same by the model. Without this, a neural network would treat each rotation as a separate instance, making learning inefficient.
Where R is a rotation and f(x) is the model’s output. This many-to-one mapping allows the model to recognize patterns across rotations, drastically reducing the amount of training data needed.
Equivariant Graph Convolutional Layer (EGCL)
The EGCL handles the relationships between nodes (particles or atoms) in the graph by processing distances and node embeddings while preserving equivariance:
-
Edge Message Calculation
This equation computes a message between two connected nodes and , where:
- and are node embeddings at layer ,
- is the squared distance between their positions,
- represents edge attributes such as charges or bond types.
-
Position Update
This updates the position of node based on the weighted sum of the relative differences in position from its neighbors. The function provides the weights, making sure that the update remains equivariant.
-
Message Aggregation
This aggregates all messages for each node , combining the information from its neighboring nodes.
-
Node Embedding Update
The node embedding is updated based on its previous state and the aggregated message . This step ensures that both node features and spatial information are updated simultaneously.
Why It Matters
- Faster Training: state-of-the-art accuracy using 1,000x less training data than other architectures, making training much more efficient in real-world applications where gathering large DFT datasets costs hundreds of millions.
- Faster inference: high accuracy with fewer layers by leveraging symmetries means improved computational scalability and the ability to predict more complex systems.
- Accuracy: by respecting real-world physics, ensuring energy and forces remain consistent under rotations or translations it becomes more reliable than regression or LLM-based models.
- Parallelizable: these models run faster by splitting message-passing and expanding the receptive field, making them more efficient on multi-GPU systems and easier to scale.
- Real-World Applicability: has tested on real-world data, predicting forces and energies in molecular dynamics for systems like water, ice, and drug molecules. Their scalability from small molecules to large periodic systems makes them versatile across domains.
Mutitask Learning
Will flush out soon. TLDR: you can learn across multiple DFT datasets with different pseudopotentials simultaneously, improving generalization for predicting properties like energies and forces with fewer data, while preserving task-specific outputs for diverse applications.